The ALPHA-MAN Project
Information about Alpha-Mannosidosis

Summary
The Disease.  The LSD alpha-Mannosidosis belongs to the glycoproteinoses affecting the glycoprotein catabolism and is characterized by immune deficiency, facial and skeletal abnormalities, hearing impairment, and intellectual disability. Alpha-Mannosidosis occurs in approximately 1 of 500,000 live births and is expected to be found in any ethnic group anywhere in the world. Alpha-Mannosidosis is caused by mutations in the MAN2B1 (LAMAN) gene encoding lysosomal alpha-Mannosidase (LAMAN). LAMAN is a lysosomal hydrolase that is responsible for cleaving alpha-mannosidic linkages during the ordered degradation of oligosaccharides. Only after degradation, can the sugars leave the lysosomes. In alpha-Mannosidosis, the deficiency of alpha-Mannosidase activity causes a block in the degradation of glycoproteins, leading to an intralysosomal accumulation of mannosyl linked oligosaccharides, that can be detected in all tissues as well as in urine and serum of patients. The progressive accumulation of oligosaccharides is somehow toxic to the cells, some types of cells are more sensitive than others. The children are often born apparently normal, and their condition worsens progressively. Since children are born clinically healthy and due to the progressive character of the disease, an early diagnosis and early initiated Enzyme Replacement Therapy could prevent neurological involvement and the fatal outcome of this disease.
The LSD alpha-Mannosidosis belongs to the glycoproteinoses affecting the glycoprotein catabolism and is characterized by immune deficiency, facial and skeletal abnormalities, hearing impairment, and intellectual disability. Alpha-Mannosidosis occurs in approximately 1 of 500,000 live births and is expected to be found in any ethnic group anywhere in the world. Alpha-Mannosidosis is caused by mutations in the MAN2B1 (LAMAN) gene encoding lysosomal alpha-Mannosidase (LAMAN). LAMAN is a lysosomal hydrolase that is responsible for cleaving alpha-mannosidic linkages during the ordered degradation of oligosaccharides. Only after degradation, can the sugars leave the lysosomes. In alpha-Mannosidosis, the deficiency of alpha-Mannosidase activity causes a block in the degradation of glycoproteins, leading to an intralysosomal accumulation of mannosyl linked oligosaccharides, that can be detected in all tissues as well as in urine and serum of patients. The progressive accumulation of oligosaccharides is somehow toxic to the cells, some types of cells are more sensitive than others. The children are often born apparently normal, and their condition worsens progressively. Since children are born clinically healthy and due to the progressive character of the disease, an early diagnosis and early initiated Enzyme Replacement Therapy could prevent neurological involvement and the fatal outcome of this disease.
Clinical Types.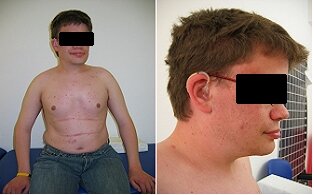 The clinical findings in alpha-Mannosidosis include a broad range of symptoms from an early lethal form to less symptomatic forms, initially diagnosed in children. Like all other LSDs, alpha-Mannosidosis lacks a genotype to phenotype correlation and even among siblings carrying the same mutation, clinical variations occur. Although the disease is a clinical spectrum three predominant clinical phenotypes of alpha Mannosidosis have been suggested. Type 1: Mild form clinically recognized after 10 years of age, without skeletal abnormalities and very slow progression. Type 2: Moderate form, clinically recognized before 10 years of age, with skeletal abnormalities, and slow progression with development of ataxia at age 20–30. Type 3: Severe form, immediately recognized, with skeletal abnormalities, and obvious progression, leading to an early death from primary central nervous system (CNS) involvement, myopathy and respiratory insuffiency. Most patients belong to clinical type 2.
The clinical findings in alpha-Mannosidosis include a broad range of symptoms from an early lethal form to less symptomatic forms, initially diagnosed in children. Like all other LSDs, alpha-Mannosidosis lacks a genotype to phenotype correlation and even among siblings carrying the same mutation, clinical variations occur. Although the disease is a clinical spectrum three predominant clinical phenotypes of alpha Mannosidosis have been suggested. Type 1: Mild form clinically recognized after 10 years of age, without skeletal abnormalities and very slow progression. Type 2: Moderate form, clinically recognized before 10 years of age, with skeletal abnormalities, and slow progression with development of ataxia at age 20–30. Type 3: Severe form, immediately recognized, with skeletal abnormalities, and obvious progression, leading to an early death from primary central nervous system (CNS) involvement, myopathy and respiratory insuffiency. Most patients belong to clinical type 2.
Diagnosis. 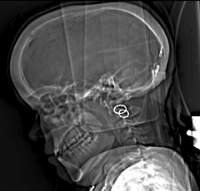 Alpha-Mannosidosis is suspected in individuals with mental retardation, hearing loss, ataxia, skeletal abnormalities and coarse features. Diagnosis occurs by biochemical testing of the alpha-Mannosidase activity in leukocytes, detection of vacuoles in lymphocytes from peripheral blood and detection of mannose-rich oligosaccharides in urine and serum of patients. Molecular genetic testing of the MAN2B1 gene and identification of two disease-causing alleles confirm the diagnosis. The alpha-Mannosidosis database, that has been developed within the HUE-MAN project (http://www.amamutdb.no) and that contains all clinical and biochemical data about alpha-Mannosidosis, will be very helpful in the future diagnosis of alpha-Mannosidosis patients, since the database is now easily accessible on the internet and will help parents and clinicians to find the right information about this rare disease, registered clinical sites, therapeutic options and family networks.
Alpha-Mannosidosis is suspected in individuals with mental retardation, hearing loss, ataxia, skeletal abnormalities and coarse features. Diagnosis occurs by biochemical testing of the alpha-Mannosidase activity in leukocytes, detection of vacuoles in lymphocytes from peripheral blood and detection of mannose-rich oligosaccharides in urine and serum of patients. Molecular genetic testing of the MAN2B1 gene and identification of two disease-causing alleles confirm the diagnosis. The alpha-Mannosidosis database, that has been developed within the HUE-MAN project (http://www.amamutdb.no) and that contains all clinical and biochemical data about alpha-Mannosidosis, will be very helpful in the future diagnosis of alpha-Mannosidosis patients, since the database is now easily accessible on the internet and will help parents and clinicians to find the right information about this rare disease, registered clinical sites, therapeutic options and family networks.